
Computational vibrational spectroscopy of molecule–surface interactions: what is still difficult and what can be done about it
 *a
and
Manabu
Ihara
a
*a
and
Manabu
Ihara
a
Abstract
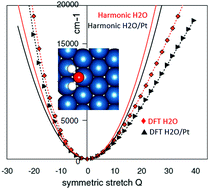
Interactions of molecules with solid surfaces are responsible for key functionalities in a range of currently actively pursued technologies, including heterogeneous catalysis for synthesis or decomposition of molecules, sensitization, surface functionalization and molecular doping, etc. Modeling of such interactions is important, in particular, for the assignment of species and assignment and design of reaction pathways and ultimately for rational design of better functional materials for a range of applications. Key types of calculations involve calculations of adsorption structures and energies, electronic structures, charge transport, vibrational and optical spectra, and reaction dynamics. While some of these calculations are routinely doable for small-size models, other types of calculations, including anharmonic vibrational spectroscopy, accurate optical spectroscopy, quantum reaction dynamics, and calculations on large systems, are still too difficult to be routinely doable. In this Perspective, we specifically focus on issues related to computing accurate vibrational spectra including quantum effects and anharmonicity and coupling of key degrees of freedom. Vibrational spectroscopies are widely used for species assignment on surfaces but computations of vibrational spectra are still dominated by the harmonic approximation. We discuss approaches that can make accurate quantum anharmonic computational spectroscopy easier and enable its wider deployment in applications. We describe advantages and disadvantages of different techniques including perturbation theory, variational, vibrational self-consistent field and vibrational configuration interaction, as well as collocation which we argue has significant potential in this application, allowing computing accurate spectra directly from non-expensive ab initio data and with modest CPU cost. Examples of applications of anharmonic techniques to molecule–surface systems are given.
- This article is part of the themed collections: 2022 PCCP HOT Articles and PCCP Reviews
 Please wait while we load your content...
Please wait while we load your content...

