
Internal conversion and intersystem crossing dynamics based on coupled potential energy surfaces with full geometry-dependent spin–orbit and derivative couplings. Nonadiabatic photodissociation dynamics of NH3(A) leading to the NH(X3Σ−, a1Δ) + H2 channel†

Abstract
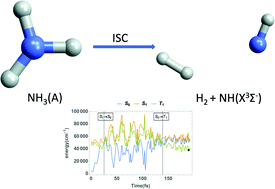
We simulate the photodissociation of NH3 originating from its first excited singlet state S1 into the NH2 + H (radical) and NH + H2 (molecular) channels. The states considered are the ground singlet state S0, the first excited singlet state S1 and the lowest-lying triplet state T1, which permit for the first time a uniform treatment of the internal conversion and intersystem crossing. The simulations are based on a diabatic potential energy matrix (DPEM) of S0, S1 coupled by a conical intersection seam, as well as a potential energy surface (PES) for T1 coupled by spin-orbit coupling (SOC) to the two singlet states. The DPEM and PES are fitted to ab initio electronic structure data (ESD) including energies, energy gradients, and derivative couplings. The DPEM also defines an adiabatic to diabatic state (AtD) transformation, which is used to transform the singular adiabatic SOC into a smooth function of the nuclear coordinates in the diabatic representation, allowing the diabatic SOC to be fit to an analytical functional form. ESD and SOC data obtained from these surfaces can serve as input for either quantum or semi-classical characterization of the nonadiabatic dynamics. Using the SHARC suite of programs, nonadiabatic simulations based on over 40 000 semi-classical trajectories assess the convergence of our results. The production of NH + H2 is not direct, but is only achieved through a quasi-statistical dissociation mechanism after internal conversion to the ground electronic state. This leads to a much lower yield comparing with the main NH2 + H channel. The NH(X3Σ_) radical produced through the intersystem crossing from S0 to T1 is rare (∼0.2%) compared to NH(a1Δ) due to the process being spin forbidden.
 Please wait while we load your content...
Please wait while we load your content...

