
A computational study of direct CO2 hydrogenation to methanol on Pd surfaces†

Abstract
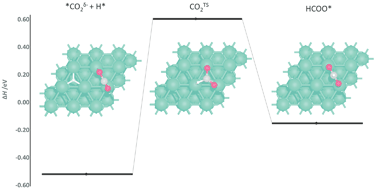
The reaction mechanism of direct CO2 hydrogenation to methanol is investigated in detail on Pd (111), (100) and (110) surfaces using density functional theory (DFT), supporting investigations into emergent Pd-based catalysts. Hydrogen adsorption and surface mobility are firstly considered, with high-coordination surface sites having the largest adsorption energy and being connected by diffusion channels with low energy barriers. Surface chemisorption of CO2, forming a partially charged CO2δ−, is weakly endothermic on a Pd (111) whilst slightly exothermic on Pd (100) and (110), with adsorption enthalpies of 0.09, −0.09 and −0.19 eV, respectively; the low stability of CO2δ− on the Pd (111) surface is attributed to negative charge accumulating on the surface Pd atoms that interact directly with the CO2δ− adsorbate. Detailed consideration for sequential hydrogenation of the CO2 shows that HCOOH hydrogenation to H2COOH would be the rate determining step in the conversion to methanol, for all surfaces, with activation barriers of 1.41, 1.51, and 0.84 eV on Pd (111), (100) and (110) facets, respectively. The Pd (110) surface exhibits overall lower activation energies than the most studied Pd (111) and (100) surfaces, and therefore should be considered in more detail in future Pd catalytic studies.
 Please wait while we load your content...
Please wait while we load your content...

