
Computational prediction of heteromeric protein complex disassembly order using hybrid Monte Carlo/molecular dynamics simulation†
 *a
and
Shigenori
Tanaka
*a
*a
and
Shigenori
Tanaka
*a
Abstract
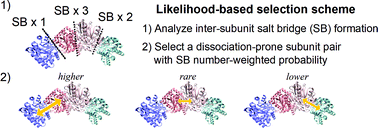
The physicochemical entities comprising the biological phenomena in the cell form a network of biochemical reactions and the activity of such a network is regulated by multimeric protein complexes. Mass spectroscopy (MS) experiments and multimeric protein docking simulations based on structural bioinformatics techniques have revealed the molecular-level stoichiometry and static configuration of subcomplexes in their bound forms, thus revealing the subcomplex population and formation orders. Meanwhile, these methodologies are not designed to straightforwardly examine the temporal dynamics of multimeric protein assembly and disassembly, essential physicochemical properties to understand the functional expression mechanisms of proteins in the biological environment. To address this problem, we have developed an atomistic simulation in the framework of the hybrid Monte Carlo/molecular dynamics (hMC/MD) method and succeeded in observing the disassembly of a homomeric pentamer of the serum amyloid P component protein in an experimentally consistent order. In this study, we improved the hMC/MD method to examine the disassembly processes of the tryptophan synthase tetramer, a paradigmatic heteromeric protein complex in MS studies. We employed the likelihood-based selection scheme to determine a dissociation-prone subunit pair at every hMC/MD simulation cycle and achieved highly reliable predictions of the disassembly orders without a priori knowledge of the MS experiments and structural bioinformatics simulations. The success rate for the experimentally-observed disassembly order is over 0.9. We similarly succeeded in reliable predictions for three other tetrameric protein complexes. These achievements indicate the potential applicability of our hMC/MD approach as a general-purpose methodology to obtain microscopic and physicochemical insights into multimeric protein complex formation.
 Please wait while we load your content...
Please wait while we load your content...

