
DLSSAffinity: protein–ligand binding affinity prediction via a deep learning model†
 *a
Yunjie
Zhao
*b
*a
Yunjie
Zhao
*b
Abstract
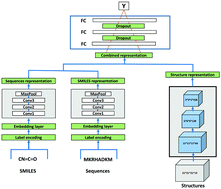
Evaluating the protein–ligand binding affinity is a substantial part of the computer-aided drug discovery process. Most of the proposed computational methods predict protein–ligand binding affinity using either limited full-length protein 3D structures or simple full-length protein sequences as the input features. Thus, protein–ligand binding affinity prediction remains a fundamental challenge in drug discovery. In this study, we proposed a novel deep learning-based approach, DLSSAffinity, to accurately predict the protein–ligand binding affinity. Unlike the existing methods, DLSSAffinity uses the pocket–ligand structural pairs as the local information to predict short-range direct interactions. Besides, DLSSAffinity also uses the full-length protein sequence and ligand SMILES as the global information to predict long-range indirect interactions. We tested DLSSAffinity on the PDBbind benchmark. The results showed that DLSSAffinity achieves Pearson's R = 0.79, RMSE = 1.40, and SD = 1.35 on the test set. Comparing DLSSAffinity with the existing state-of-the-art deep learning-based binding affinity prediction methods, the DLSSAffinity model outperforms other models. These results demonstrate that combining global sequence and local structure information as the input features of a deep learning model can improve the accuracy of protein–ligand binding affinity prediction.
 Please wait while we load your content...
Please wait while we load your content...

