
Desorption trends of small alcohols and the disruption of intermolecular interactions at defect sites on Au(111)†

Abstract
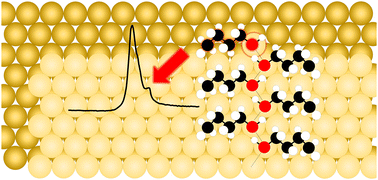
Gold-based catalysts have received tremendous attention as supports and nanoparticles for heterogeneous catalysis, in part due to the ability of nanoscale Au to catalyze reactions at low temperatures in oxidative environments. Surface defects are known active sites for low temperature Au chemistry, so a full understanding of the interplay between intermolecular interactions and surface morphology is essential to an advanced understanding of catalytic behavior and efficiency. In a systematic study to better understand the adsorption and intermolecular behavior of small alcohols (C1–C4) on Au(111) defect sites, coverage studies of methanol, ethanol, 1-propanol, 1-butanol, 2-butanol, and isobutanol have been conducted on Au(111) using ultrahigh vacuum temperature programmed desorption (UHV-TPD). These small alcohols molecularly adsorb on the Au(111) surface and high resolution experiments reveal distinct terrace, step edge, and kink adsorption features for each molecule. The hydrogen-bonded (H-bonded) networks of small alcohols on Au(111), except for 1-butanol and isobutanol, have been previously imaged on the molecular level at low temperatures by scanning tunneling microscopy. Primary C1–C3 alcohols exhibit planar H-bonded long extended zigzag chain networks while 2-butanol arranges in tetramer clusters of H-bonded molecules due to steric hindrance inhibiting the proximity of molecules on Au(111). Herein, the desorption energy of small primary alcohols was shown to trend linearly with increasing C1–C4 carbon chain length, indicating that the H-bonded molecular packing of 1-butanol resembles that of methanol, ethanol, and 1-propanol, while isobutanol and 2-butanol deviate from the trend. Butanol isomer studies allow the prediction of isobutanol long extended chains in contrast to tetramers. The distinction between the desorption of butanol isomers highlights the role of intermolecular interactions due to the difference in molecular packing structures on Au(111). Furthermore, by studying the energetics of terrace H-bonded networks in comparison with molecular adsorption at undercoordinated step edge and kink defect sites, it is shown that the contribution of stabilizing van der Waals forces to the overall adsorption energy is less for small alcohols adsorbed at kink sites (3.1 kJ mol−1 per CH2) and similar for those adsorbed at step edge (4.8 kJ mol−1 per CH2) and Au terrace sites (4.9 kJ mol−1 per CH2).
 Please wait while we load your content...
Please wait while we load your content...

