
Molecular insight into the role of zeolite lattice constraints on methane activation over the Cu–O–Cu active site†

Abstract
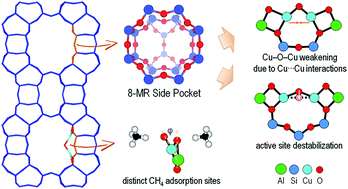
Understanding the factors that influence the activity of a catalyst toward CH4 activation is of high importance for tuning the catalyst performance or designing new, better catalysts. Here, we performed a set of density functional theory (DFT) calculations on the H–CH3 bond cleavage over the Cu–O–Cu active site in the MOR zeolite with various Al-pair arrangements to obtain molecular insight into the structure–activity relation and clarify key parameters that define the Cu–O–Cu reactivity toward CH4. We found that weakening of the Cu–O–Cu bond during CH4 activation is crucial for determining the O–H bond strength and thus the Cu–O–Cu reactivity. In this regard, the zeolite lattice constraints are found to play a significant role as, on the one hand, it strengthens the Cu⋯Cu interaction and consequently weakens the Cu–O–Cu bonds and, on the other hand, it forces the Cu–O–Cu bond elongation process to destabilize the active site structure. The non-planar Cu–O–Cu geometry, due to lattice constraints, is also found to make the CH4 adsorption site, whether positioned closer to the μ-O or the Cu atom, crucial in determining the C–H activation product, i.e., a ˙CH3 radical or a Cu2–CH3− ligand.
- This article is part of the themed collection: 2022 PCCP HOT Articles
 Please wait while we load your content...
Please wait while we load your content...

