
Molecular dynamics of liquid–liquid equilibrium and interfacial properties of aqueous solutions of methyl esters

Abstract
In this work, the liquid–liquid phase equilibria and interfacial properties of methyl ester + water binary mixtures are determined at atmospheric pressure and from 278 to 358 K combining the direct coexistence technique and molecular dynamics simulations. Methyl esters are modelled using new parametrizations based on the united atom TraPPE model force field proposed recently by us [E. Feria, J. Algaba, J. M. Míguez, A. Mejía, P. Gómez-Álvarez and F. J. Blas, Phys. Chem. Chem. Phys., 2019, 22, 4974–4983] that are able to predict the vapour–liquid interfacial properties of pure methyl esters with high accuracy. In the case of water, we consider the well-known TIP4P/2005 model, the most popular rigid and non-polarizable model to describe the interfacial properties of pure water. The simulations are performed using the direct coexistence technique in the isothermal–isobaric or NPz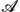 T ensemble in combination with molecular dynamics. We obtain density profiles, temperature–densities and temperature–composition projections of the phase diagrams, and interfacial tensions. The liquid–liquid interfacial tension is calculated from the normal and tangential components of the pressure tensor according to the mechanical virial route. We pay attention particularly to the ability of the molecular models in predicting the experimental behavior of the systems. Simulation results are able to account for the liquid–liquid phase equilibria of these binary mixtures, in good agreement with the experimental data taken from the literature. Unfortunately, experimental values for interfacial tensions are substantially overestimated by predictions from computer simulations in all cases. To our knowledge, this is the first time that the liquid–liquid phase equilibrium and interfacial properties of methyl ester + water mixtures have been predicted from computer simulations.
T ensemble in combination with molecular dynamics. We obtain density profiles, temperature–densities and temperature–composition projections of the phase diagrams, and interfacial tensions. The liquid–liquid interfacial tension is calculated from the normal and tangential components of the pressure tensor according to the mechanical virial route. We pay attention particularly to the ability of the molecular models in predicting the experimental behavior of the systems. Simulation results are able to account for the liquid–liquid phase equilibria of these binary mixtures, in good agreement with the experimental data taken from the literature. Unfortunately, experimental values for interfacial tensions are substantially overestimated by predictions from computer simulations in all cases. To our knowledge, this is the first time that the liquid–liquid phase equilibrium and interfacial properties of methyl ester + water mixtures have been predicted from computer simulations.

 Please wait while we load your content...
Please wait while we load your content...

