
Variational quantum eigensolver simulations with the multireference unitary coupled cluster ansatz: a case study of the C2v quasi-reaction pathway of beryllium insertion into a H2 molecule†
 *abc
Yuji
Mochizuki
ef
*abc
Yuji
Mochizuki
ef
Abstract
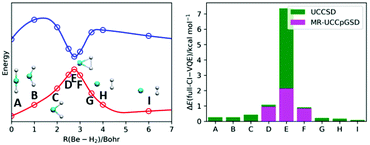
Variational quantum eigensolver (VQE)-based quantum chemical calculations have been extensively studied as a computational model using noisy intermediate-scale quantum devices. The VQE uses a parametrized quantum circuit defined through an “ansatz” to generate approximated wave functions, and the appropriate choice of an ansatz is the most important step. Because most chemistry problems focus on the energy difference between two electronic states or structures, calculating the total energies in different molecular structures with the same accuracy is essential to correctly understand chemistry and chemical processes. In this context, the development of ansatzes that are capable of describing electronic structures of strongly correlated systems accurately is an important task. Here we applied a conventional unitary coupled cluster (UCC) and a newly developed multireference unitary coupled cluster with partially generalized singles and doubles (MR-UCCpGSD) ansatzes to the quasi-reaction pathway of Be insertion into H2, LiH molecule under covalent bond dissociation, and a rectangular tetra-hydrogen cluster known as a P4 cluster; these are representative systems in which the static electron correlation effect is prominent. Our numerical simulations revealed that the UCCSD ansatz exhibits extremely slow convergence behaviour around the point where an avoided crossing occurs in the Be + H2 → BeH2 reaction pathway, resulting in a large discrepancy of the simulated VQE energy from the full-configuration interaction (full-CI) value. By contrast, the MR-UCCpGSD ansatz can give more reliable results with respect to total energy and the overlap with the full-CI solution, insisting the importance of multiconfigurational treatments in the calculations of strongly correlated systems. The MR-UCCpGSD ansatz allows us to compute the energy with the same accuracy regardless of the strength of multiconfigurational character, which is an essential property to discuss energy differences of various molecular systems.
 Please wait while we load your content...
Please wait while we load your content...

