
Machine learning and semi-empirical calculations: a synergistic approach to rapid, accurate, and mechanism-based reaction barrier prediction†
 a
and
Matthew N.
Grayson
*a
a
and
Matthew N.
Grayson
*a
Abstract
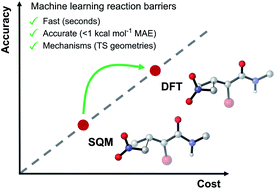
Modern QM modelling methods, such as DFT, have provided detailed mechanistic insights into countless reactions. However, their computational cost inhibits their ability to rapidly screen large numbers of substrates and catalysts in reaction discovery. For a C–C bond forming nitro-Michael addition, we introduce a synergistic semi-empirical quantum mechanical (SQM) and machine learning (ML) approach that allows the prediction of DFT-quality reaction barriers in minutes, even on a standard laptop using widely available modelling software. Mean absolute errors (MAEs) are obtained that are below the accepted chemical accuracy threshold of 1 kcal mol−1 and substantially better than SQM methods without ML correction (5.71 kcal mol−1). Predictive power is shown to hold when the ML models are applied to an unseen set of compounds from the toxicology literature. Mechanistic insight is also achieved via the generation of full SQM transition state (TS) structures which are found to be very good approximations for the DFT-level geometries, revealing important steric interactions in some TSs. This combination of speed, accuracy, and mechanistic insight is unprecedented; current ML barrier models compromise on at least one of these important criteria.
 Please wait while we load your content...
Please wait while we load your content...

