
A machine learning protocol for revealing ion transport mechanisms from dynamic NMR shifts in paramagnetic battery materials†

Abstract
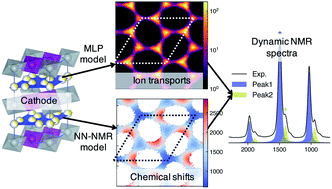
Solid-state nuclear magnetic resonance (ssNMR) provides local environments and dynamic fingerprints of alkali ions in paramagnetic battery materials. Linking the local ionic environments and NMR signals requires expensive first-principles computational tools that have been developed for over a decade. Nevertheless, the assignment of the dynamic NMR spectra of high-rate battery materials is still challenging because the local structures and dynamic information of alkali ions are highly correlated and difficult to acquire. Herein, we develop a novel machine learning (ML) protocol that could not only quickly sample atomic configurations but also predict chemical shifts efficiently, which enables us to calculate dynamic NMR shifts with the accuracy of density functional theory (DFT). Using structurally well-defined P2-type Na2/3(Mg1/3Mn2/3)O2 as an example, we validate the ML protocol and show the significance of dynamic effects on chemical shifts. Moreover, with the protocol, it is demonstrated that the two experimental 23Na shifts (1406 and 1493 ppm) of P2-type Na2/3(Ni1/3Mn2/3)O2 originate from two stacking sequences of transition metal (TM) layers for the first time, which correspond to space groups P63/mcm and P6322, respectively. This ML protocol could help to correlate dynamic ssNMR spectra with the local structures and fast transport of alkali ions and is expected to be applicable to a wide range of fast dynamic systems.
 Please wait while we load your content...
Please wait while we load your content...

