
A first principles examination of phosphorescence

Abstract
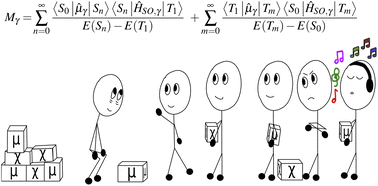
This paper explores phosphorescence from a first principles standpoint, and examines the intricacies involved in calculating the spin-forbidden T1 → S0 transition dipole moment, to highlight that the mechanism is not as complicated to compute as it seems. Using gas phase acridine as a case study, we break down the formalism required to compute the phosphorescent spectra within both the Franck–Condon and Herzberg–Teller regimes by coupling the first triplet excited state up to the S4 and T4 states. Despite the first singlet excited state appearing as an Lb state and not of nπ* character, the second order corrected rate constant was found to be 0.402 s−1, comparing well with experimental phosphorescent lifetimes of acridine derivatives. In showing only certain states are required to accurately describe the matrix elements as well as how to find these states, our calculations suggest that the nπ* state only weakly couples to the T1 state. This suggest its importance hinges on its ability to quench fluorescence and exalt non-radiative mechanisms rather than its contribution to the transition dipole moment. A followup investigation into the T1 → S0 transition dipole moment's growth as a function of its coupling to other electronic states highlights that terms dominating the matrix element arise entirely from the inclusion of states with strong spin–orbit coupling terms. This means that while the expansion of the transition dipole moment can extend to include an infinite number of electronic states, only certain states need to be included.
 Please wait while we load your content...
Please wait while we load your content...

