
Kinetics and mechanism of sequential ring methyl C–H activation in cyclopentadienyl rhodium(iii) complexes†
 ‡a
Samya
Banerjee,
‡*ab
Juliusz A.
Wolny,
‡*c
Cinzia
Imberti,
a
Edward C.
Lant,
a
Marc
Walker,
d
Volker
Schünemann
c
and
Peter J.
Sadler
*a
‡a
Samya
Banerjee,
‡*ab
Juliusz A.
Wolny,
‡*c
Cinzia
Imberti,
a
Edward C.
Lant,
a
Marc
Walker,
d
Volker
Schünemann
c
and
Peter J.
Sadler
*a
Abstract
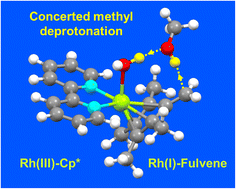
We have studied activation of the methyl C–H bonds in the cyclopentadienyl ligands of half-sandwich Rh(III) complexes [η5-CpXRh(N,N′)Cl]+ by observing the dependence of sequential H/D exchange on variations in CpX = Cp* (complexes 1 and 2), Me4PhCp (CpXPh, 3) or Me4PhPhCp (CpXPhPh, 4), and chelated ligand N,N′ (bpy, 1; phen, 2–4). H/D exchange was fastest in d4-MeOD (t1/2 = 10 min, 37 °C, complex 1), no H/D exchange was observed in DMSO/D2O, and d4-MeOD enhanced the rate in CD3CN. The proposed Rh(I)–fulvene intermediate was trapped by [4 + 2] Diels–Alder reactions with conjugated dienes and characterized. The Rh(I) oxidation state was confirmed by X-ray photoelectron spectroscopy (XPS). Influence of solvent on the mechanisms of activation and Diels–Alder adduct formation was modelled using DFT calculations with the CAM-B3LYP functional and CEP-31 g basis set, and influence on the reaction profile of the dimiine ligand and phenyl substituent using the larger qzvp basis set. The Rh(III)–OH intemediate is stabilised by H-bonding with methanol and a Cp* CH3 hydrogen. The Rh(I)(Me4fulvene) species, stabilised by interaction of methanol with a coordinated water, again by two H-bonds H2O–HOMe (1.49 Å) and fulvene CH2 (1.94 Å), arises from synchronous transfer of the methanol OH proton to a Rh(III)–OH ligand and Cp* methyl hydrogen to the methanol oxygen. Additionally, the observed trend in catalytic activity for complexes 1–4 was reproduced by DFT calculations. These complexes form a novel class of catalytic molecular motors with a tunable rate of operation that can be stalled in a given state. They provide a basis for elucidation of the effects of ligand design on the contributions of electronic, rotational and vibrational energies to each step in the reaction pathway at the atomic level, consideration of which will enhance the design principles for the next generation of molecular machines.
 Please wait while we load your content...
Please wait while we load your content...

