
Exploration and validation of force field design protocols through QM-to-MM mapping†

Abstract
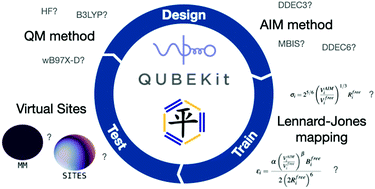
The scale of the parameter optimisation problem in traditional molecular mechanics force field construction means that design of a new force field is a long process, and sub-optimal choices made in the early stages can persist for many generations. We hypothesise that careful use of quantum mechanics to inform molecular mechanics parameter derivation (QM-to-MM mapping) should be used to significantly reduce the number of parameters that require fitting to experiment and increase the pace of force field development. Here, we design and train a collection of 15 new protocols for small, organic molecule force field derivation, and test their accuracy against experimental liquid properties. Our best performing model has only seven fitting parameters, yet achieves mean unsigned errors of just 0.031 g cm−3 and 0.69 kcal mol−1 in liquid densities and heats of vaporisation, compared to experiment. The software required to derive the designed force fields is freely available at https://github.com/qubekit/QUBEKit.
- This article is part of the themed collection: 2022 PCCP HOT Articles
 Please wait while we load your content...
Please wait while we load your content...

