
Double proton transfer in hydrated formic acid dimer: Interplay of spatial symmetry and solvent-generated force on reactivity†

Abstract
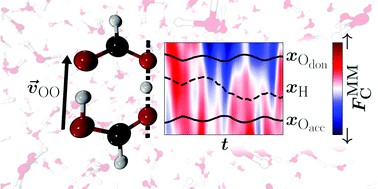
The double proton transfer (DPT) reaction in the hydrated formic acid dimer (FAD) is investigated at molecular-level detail. For this, a global and reactive machine learned (ML) potential energy surface (PES) is developed to run extensive (more than 100 ns) mixed ML/MM molecular dynamics (MD) simulations in explicit molecular mechanics (MM) solvent at MP2-quality for the solute. Simulations with fixed – as in a conventional empirical force field – and conformationally fluctuating – as available from the ML-based PES – charge models for FAD show a significant impact on the competition between DPT and dissociation of FAD into two formic acid monomers. With increasing temperature the barrier height for DPT in solution changes by about 10% (∼1 kcal mol−1) between 300 K and 600 K. The rate for DPT is largest, ∼1 ns−1, at 350 K and decreases for higher temperatures due to destabilisation and increased probability for dissociation of FAD. The water solvent is found to promote the first proton transfer by exerting a favourable solvent-induced Coulomb force along the O–H⋯O hydrogen bond whereas the second proton transfer is significantly controlled by the O–O separation and other conformational degrees of freedom. Double proton transfer in hydrated FAD is found to involve a subtle interplay and balance between structural and electrostatic factors.
 Please wait while we load your content...
Please wait while we load your content...

