
Benchmarks of the density functional tight-binding method for redox, protonation and electronic properties of quinones†

Abstract
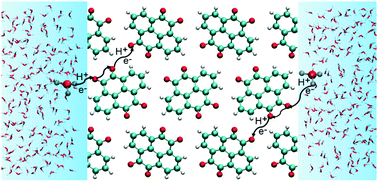
Organic materials with controllable molecular design and sustainable resources are promising electrode materials. Crystalline quinones have been investigated in a variety of rechargeable battery chemistries due to their ubiquitous nature, voltage tunability and environmental friendliness. In acidic electrolytes, quinone crystals can undergo proton-coupled electron transfer (PCET), resulting in charge storage. However, the detailed mechanism of this phenomenon remains elusive. To model PCET in crystalline quinones, force field-based methods are not viable due to variable redox states of the quinone molecules during battery operation and computationally efficient quantum mechanical methods are strongly desired. The semi-empirical density functional tight-binding (DFTB) method has been widely used to study inorganic crystalline systems and biological systems but has not been comprehensively benchmarked for studying charge transport in quinones. In this work, we benchmark the third order variant of DFTB (DFTB3) for the reduction potential of quinones in aqueous solution, energetics of proton transfer between quinones and between quinones and water, and structural and electronic properties of crystalline quinones. Our results reveal the deficiencies of the DFTB3 method in describing the proton affinity of quinones and the structural and electronic properties of crystalline quinones, and highlight the need for further development of the DFTB method for describing charge transport in crystalline quinones.
 Please wait while we load your content...
Please wait while we load your content...

