
Along the road to crystal structure prediction (CSP) of pharmaceutical-like molecules
 *a
and
Kacper
Drużbicki
ab
*a
and
Kacper
Drużbicki
ab
Abstract
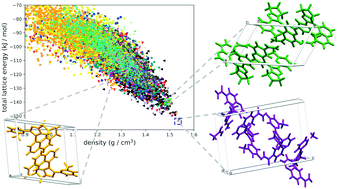
Computational methods used for predicting the crystal structures of organic compounds are mature enough to be routinely used with many rigid and semi-rigid organic molecules. The usefulness of crystal structure prediction (CSP) is recognized not only by academia but also by the pharmaceutical industry, with an ability to accelerate the polymorph screening process and minimize the risk of unanticipated late-appearing polymorphs. The challenge, however, remains when molecules with high flexibility are considered. The same applies to high-Z′ and/or multi-component crystal structures. This is because each degree of freedom causes an exponential rise in the number of structures that have to be evaluated, while often being nearly energetically degenerate. As a result, many researchers put a great deal of effort into aiding CSP of flexible and/or multi-component systems. In this work, a brief overview of the current capability of CSP methods to solve these issues is given, with a special emphasis on their application for pharmaceutical-like molecules.
- This article is part of the themed collection: 2022 Highlight article collection
 Please wait while we load your content...
Please wait while we load your content...

