
Cereblon covalent modulation through structure-based design of histidine targeting chemical probes†

Abstract
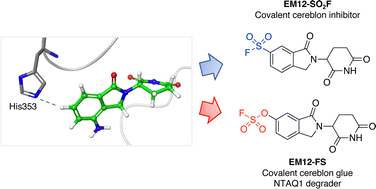
Electrophilic biocompatible warheads, particularly cysteine-reactive acrylamides, have enabled the development of covalent inhibitor drugs and chemical biology probes, but cysteine is rarely present in protein binding sites. Therefore, expansion of the list of targetable amino acid residues is required to augment the synthetic bology toolkit of site-selective protein modifications. This work describes the first rational targeting of a specific histidine residue in a protein binding site using sulfonyl exchange chemistry. Structure-based drug design was used to incorporate sulfonyl fluoride and triazole reactive groups into the isoindolinone thalidomide congener EM12 to yield potent covalent inhibitors of the cereblon E3 ubiquitin ligase complex through engagement of His353. Conversely, the fluorosulfate derivative EM12-FS labels His353, but degrades a novel neosubstrate, the protein N-terminal glutamine amidohydrolase NTAQ1, which is involved in the N-end rule pathway and DNA damage response. Targeted protein degradation using cereblon ligands has become an important new drug discovery modality and the chemical probes and covalent labeling strategy described here will broadly impact this exciting area of therapeutic research.
 Please wait while we load your content...
Please wait while we load your content...

