
Structure prediction of cyclic peptides by molecular dynamics + machine learning†
 a
Yu-Shan
Lin
*a
a
Yu-Shan
Lin
*a
Abstract
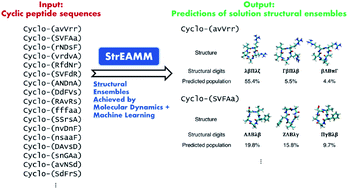
Recent computational methods have made strides in discovering well-structured cyclic peptides that preferentially populate a single conformation. However, many successful cyclic-peptide therapeutics adopt multiple conformations in solution. In fact, the chameleonic properties of some cyclic peptides are likely responsible for their high cell membrane permeability. Thus, we require the ability to predict complete structural ensembles for cyclic peptides, including the majority of cyclic peptides that have broad structural ensembles, to significantly improve our ability to rationally design cyclic-peptide therapeutics. Here, we introduce the idea of using molecular dynamics simulation results to train machine learning models to enable efficient structure prediction for cyclic peptides. Using molecular dynamics simulation results for several hundred cyclic pentapeptides as the training datasets, we developed machine-learning models that can provide molecular dynamics simulation-quality predictions of structural ensembles for all the hundreds of thousands of sequences in the entire sequence space. The prediction for each individual cyclic peptide can be made using less than 1 second of computation time. Even for the most challenging classes of poorly structured cyclic peptides with broad conformational ensembles, our predictions were similar to those one would normally obtain only after running multiple days of explicit-solvent molecular dynamics simulations. The resulting method, termed StrEAMM (Structural Ensembles Achieved by Molecular Dynamics and Machine Learning), is the first technique capable of efficiently predicting complete structural ensembles of cyclic peptides without relying on additional molecular dynamics simulations, constituting a seven-order-of-magnitude improvement in speed while retaining the same accuracy as explicit-solvent simulations.
 Please wait while we load your content...
Please wait while we load your content...

