
Identifying the true origins of selectivity in chiral phosphoric acid catalyzed N-acyl-azetidine desymmetrizations†

Abstract
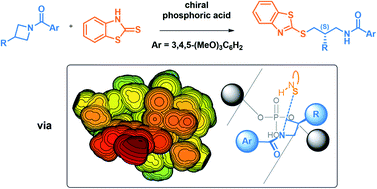
The first catalytic intermolecular desymmetrization of azetidines was reported by Sun and coworkers in 2015 using a BINOL-derived phosphoric acid catalyst (J. Am. Chem. Soc. 2015, 137, 5895–5898). To uncover the mechanism of the reaction and the origins of the high enantioselectivity, Density Functional Theory (DFT) calculations were performed at the B97D3/6-311+G(2d,2p)/SMD(toluene)//B97D3/6-31G(d,p)/CPCM(toluene) level of theory. Comparison of four possible activation modes confirms that this reaction proceeds through the bifunctional activation of the azetidine nitrogen and the thione tautomer of the 2-mercaptobenzothiazole nucleophile. Upon thorough conformational sampling of the enantiodetermining transition structures (TSs), a free energy difference of 2.0 kcal mol−1 is obtained, accurately reproducing the experimentally measured 88% e.e. at 80 °C. This energy difference is due to both decreased distortion and increased non-covalent interactions in the pro-(S) TS. To uncover the true origins of selectivity, the TSs optimized with the full catalyst were compared to those optimized with a model catalyst through steric maps. It is found that the arrangements displayed by the substrates are controlled by strict primary orbital interaction requirements at the transition complex, and their ability to fit into the catalyst pocket drives the selectivity. A general model of selectivity for phosphoric acid-catalyzed azetidine desymmetrizations is proposed, which is based on the preference of the nucleophile and benzoyl group to occupy empty quadrants of the chiral catalyst pocket.
 Please wait while we load your content...
Please wait while we load your content...

