
Structure-based de novo drug design using 3D deep generative models†

Abstract
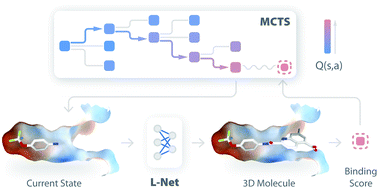
Deep generative models are attracting much attention in the field of de novo molecule design. Compared to traditional methods, deep generative models can be trained in a fully data-driven way with little requirement for expert knowledge. Although many models have been developed to generate 1D and 2D molecular structures, 3D molecule generation is less explored, and the direct design of drug-like molecules inside target binding sites remains challenging. In this work, we introduce DeepLigBuilder, a novel deep learning-based method for de novo drug design that generates 3D molecular structures in the binding sites of target proteins. We first developed Ligand Neural Network (L-Net), a novel graph generative model for the end-to-end design of chemically and conformationally valid 3D molecules with high drug-likeness. Then, we combined L-Net with Monte Carlo tree search to perform structure-based de novo drug design tasks. In the case study of inhibitor design for the main protease of SARS-CoV-2, DeepLigBuilder suggested a list of drug-like compounds with novel chemical structures, high predicted affinity, and similar binding features to those of known inhibitors. The current version of L-Net was trained on drug-like compounds from ChEMBL, which could be easily extended to other molecular datasets with desired properties based on users' demands and applied in functional molecule generation. Merging deep generative models with atomic-level interaction evaluation, DeepLigBuilder provides a state-of-the-art model for structure-based de novo drug design and lead optimization.
- This article is part of the themed collections: 2021 Chemical Science HOT Article Collection and In celebration of Chinese New Year, 2022
 Please wait while we load your content...
Please wait while we load your content...

