
Predicting chemical shifts with graph neural networks†

Abstract
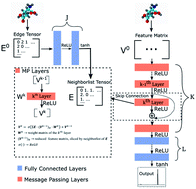
Inferring molecular structure from Nuclear Magnetic Resonance (NMR) measurements requires an accurate forward model that can predict chemical shifts from 3D structure. Current forward models are limited to specific molecules like proteins and state-of-the-art models are not differentiable. Thus they cannot be used with gradient methods like biased molecular dynamics. Here we use graph neural networks (GNNs) for NMR chemical shift prediction. Our GNN can model chemical shifts accurately and capture important phenomena like hydrogen bonding induced downfield shift between multiple proteins, secondary structure effects, and predict shifts of organic molecules. Previous empirical NMR models of protein NMR have relied on careful feature engineering with domain expertise. These GNNs are trained from data alone with no feature engineering yet are as accurate and can work on arbitrary molecular structures. The models are also efficient, able to compute one million chemical shifts in about 5 seconds. This work enables a new category of NMR models that have multiple interacting types of macromolecules.
 Please wait while we load your content...
Please wait while we load your content...

