
The role of hypervalent iodine(iii) reagents in promoting alkoxylation of unactivated C(sp3)–H bonds catalyzed by palladium(ii) complexes†

Abstract
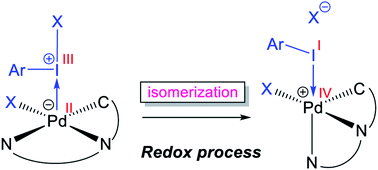
Although Pd(OAc)2-catalysed alkoxylation of the C(sp3)–H bonds mediated by hypervalent iodine(III) reagents (ArIX2) has been developed by several prominent researchers, there is no clear mechanism yet for such crucial transformations. In this study, we shed light on this important issue with the aid of the density functional theory (DFT) calculations for alkoxylation of butyramide derivatives. We found that the previously proposed mechanism in the literature is not consistent with the experimental observations and thus cannot be operating. The calculations allowed us to discover an unprecedented mechanism composed of four main steps as follows: (i) activation of the C(sp3)–H bond, (ii) oxidative addition, (iii) reductive elimination and (iv) regeneration of the active catalyst. After completion of step (i) via the CMD mechanism, the oxidative addition commences with an X ligand transfer from the iodine(III) reagent (ArIX2) to Pd(II) to form a square pyramidal complex in which an iodonium occupies the apical position. Interestingly, a simple isomerization of the resultant five-coordinate complex triggers the Pd(II) oxidation. Accordingly, the movement of the ligand trans to the Pd–C(sp3) bond to the apical position promotes the electron transfer from Pd(II) to iodine(III), resulting in the reduction of iodine(III) concomitant with the ejection of the second X ligand as a free anion. The ensuing Pd(IV) complex then undergoes the C–O reductive elimination by nucleophilic attack of the solvent (alcohol) on the sp3 carbon via an outer-sphere SN2 mechanism assisted by the X− anion. Noteworthy, starting from the five coordinate complex, the oxidative addition and reductive elimination processes occur with a very low activation barrier (ΔG‡ 0–6 kcal mol−1). The strong coordination of the alkoxylated product to the Pd(II) centre causes the regeneration of the active catalyst, i.e. step (iv), to be considerably endergonic, leading to subsequent catalytic cycles to proceed with a much higher activation barrier than the first cycle. We also found that although, in most cases, the alkoxylation reactions proceed via a Pd(II)–Pd(IV)–Pd(II) catalytic cycle, the other alternative in which the oxidation state of the Pd(II) centre remains unchanged during the catalysis could be operative, depending on the nature of the organic substrate.
 Please wait while we load your content...
Please wait while we load your content...

