
Rational design of thioamide peptides as selective inhibitors of cysteine protease cathepsin L†

Abstract
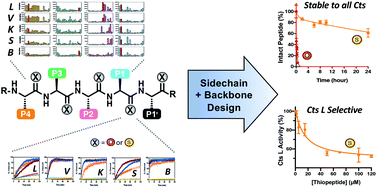
Aberrant levels of cathepsin L (Cts L), a ubiquitously expressed endosomal cysteine protease, have been implicated in many diseases such as cancer and diabetes. Significantly, Cts L has been identified as a potential target for the treatment of COVID-19 due to its recently unveiled critical role in SARS-CoV-2 entry into the host cells. However, there are currently no clinically approved specific inhibitors of Cts L, as it is often challenging to obtain specificity against the many highly homologous cathepsin family cysteine proteases. Peptide-based agents are often promising protease inhibitors as they offer high selectivity and potency, but unfortunately are subject to degradation in vivo. Thioamide substitution, a single-atom O-to-S modification in the peptide backbone, has been shown to improve the proteolytic stability of peptides addressing this issue. Utilizing this approach, we demonstrate herein that good peptidyl substrates can be converted into sub-micromolar inhibitors of Cts L by a single thioamide substitution in the peptide backbone. We have designed and scanned several thioamide stabilized peptide scaffolds, in which one peptide, RS1A, was stabilized against proteolysis by all five cathepsins (Cts L, Cts V, Cts K, Cts S, and Cts B) while inhibiting Cts L with >25-fold specificity against the other cathepsins. We further showed that this stabilized RS1A peptide could inhibit Cts L in human liver carcinoma lysates (IC50 = 19 μM). Our study demonstrates that one can rationally design a stabilized, specific peptidyl protease inhibitor by strategic placement of a thioamide and reaffirms the place of this single-atom modification in the toolbox of peptide-based rational drug design.
 Please wait while we load your content...
Please wait while we load your content...

