
Reaction-based machine learning representations for predicting the enantioselectivity of organocatalysts†
 ‡a
Raimon
Fabregat,
‡a
Rubén
Laplaza,
ab
Sinjini
Bhattacharjee,
ac
Matthew D.
Wodrich
ab
and
Clemence
Corminboeuf
*abd
‡a
Raimon
Fabregat,
‡a
Rubén
Laplaza,
ab
Sinjini
Bhattacharjee,
ac
Matthew D.
Wodrich
ab
and
Clemence
Corminboeuf
*abd
Abstract
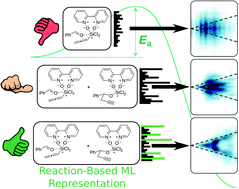
Hundreds of catalytic methods are developed each year to meet the demand for high-purity chiral compounds. The computational design of enantioselective organocatalysts remains a significant challenge, as catalysts are typically discovered through experimental screening. Recent advances in combining quantum chemical computations and machine learning (ML) hold great potential to propel the next leap forward in asymmetric catalysis. Within the context of quantum chemical machine learning (QML, or atomistic ML), the ML representations used to encode the three-dimensional structure of molecules and evaluate their similarity cannot easily capture the subtle energy differences that govern enantioselectivity. Here, we present a general strategy for improving molecular representations within an atomistic machine learning model to predict the DFT-computed enantiomeric excess of asymmetric propargylation organocatalysts solely from the structure of catalytic cycle intermediates. Mean absolute errors as low as 0.25 kcal mol−1 were achieved in predictions of the activation energy with respect to DFT computations. By virtue of its design, this strategy is generalisable to other ML models, to experimental data and to any catalytic asymmetric reaction, enabling the rapid screening of structurally diverse organocatalysts from available structural information.
- This article is part of the themed collections: 2021 Chemical Science HOT Article Collection, 2021 Nobel Prize in Chemistry – Asymmetric Organocatalysis and Editor’s Choice – Graeme Day
 Please wait while we load your content...
Please wait while we load your content...

