
Statistical analysis of C–H activation by oxo complexes supports diverse thermodynamic control over reactivity†

Abstract
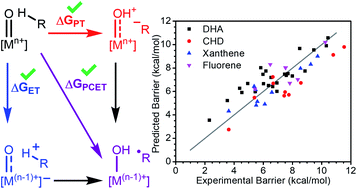
Transition metal oxo species are key intermediates for the activation of strong C–H bonds. As such, there has been interest in understanding which structural or electronic parameters of metal oxo complexes determine their reactivity. Factors such as ground state thermodynamics, spin state, steric environment, oxygen radical character, and asynchronicity have all been cited as key contributors, yet there is no consensus on when each of these parameters is significant or the relative magnitude of their effects. Herein, we present a thorough statistical analysis of parameters that have been proposed to influence transition metal oxo mediated C–H activation. We used density functional theory (DFT) to compute parameters for transition metal oxo complexes and analyzed their ability to explain and predict an extensive data set of experimentally determined reaction barriers. We found that, in general, only thermodynamic parameters play a statistically significant role. Notably, however, there are independent and significant contributions from the oxidation potential and basicity of the oxo complexes which suggest a more complicated thermodynamic picture than what has been shown previously.
 Please wait while we load your content...
Please wait while we load your content...

