
How metallylenes activate small molecules†

Abstract
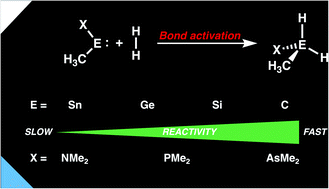
We have studied the activation of dihydrogen by metallylenes using relativistic density functional theory (DFT). Our detailed activation strain and Kohn–Sham molecular orbital analyses have quantified the physical factors behind the decreased reactivity of the metallylene on going down Group 14, from carbenes to stannylenes. Along this series, the reactivity decreases due to a worsening of the back-donation interaction between the filled lone-pair orbital of the metallylene and the σ*-orbital of H2, which, therefore, reduces the metallylene–substrate interaction and increases the reaction barrier. As the metallylene ligand is varied from nitrogen to phosphorus to arsenic a significant rate enhancement is observed for the activation of H2 due to (i) a reduced steric (Pauli) repulsion between the metallylene and the substrate; and (ii) less activation strain, as the metallylene becomes increasingly more predistorted. Using a rationally designed metallylene with an optimal Group 14 atom and ligand combination, we show that a number of small molecules (i.e. HCN, CO2, H2, NH3) may also be readily activated. For the first time, we show the ability of our H2 activated designer metallylenes to hydrogenate unsaturated hydrocarbons. The results presented herein will serve as a guide for the rational design of metallylenes toward the activation of small molecules and subsequent reactions.
 Please wait while we load your content...
Please wait while we load your content...

