
Ligand design by targeting a binding site water†

Abstract
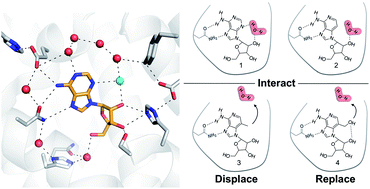
Solvent reorganization is a major driving force of protein–ligand association, but the contribution of binding site waters to ligand affinity is poorly understood. We investigated how altered interactions with a water network can influence ligand binding to a receptor. A series of ligands of the A2A adenosine receptor, which either interacted with or displaced an ordered binding site water, were studied experimentally and by molecular dynamics simulations. An analog of the endogenous ligand that was unable to hydrogen bond to the ordered water lost affinity and this activity cliff was captured by molecular dynamics simulations. Two compounds designed to displace the ordered water from the binding site were then synthesized and evaluated experimentally, leading to the discovery of an A2A agonist with nanomolar activity. Calculation of the thermodynamic profiles resulting from introducing substituents that interacted with or displaced the ordered water showed that the gain of binding affinity was enthalpy driven. Detailed analysis of the energetics and binding site hydration networks revealed that the enthalpy change was governed by contributions that are commonly neglected in structure-based drug optimization. In particular, simulations suggested that displacement of water from a binding site to the bulk solvent can lead to large energy contributions. Our findings provide insights into the molecular driving forces of protein–ligand binding and strategies for rational drug design.
 Please wait while we load your content...
Please wait while we load your content...

