
Mechanistic insights into the insertion and addition reactions of group 13 analogues of the six-membered N-heterocyclic carbenes: interplay of electrophilicity, basicity, and aromaticity governing the reactivity†
 *ab
*ab
Abstract
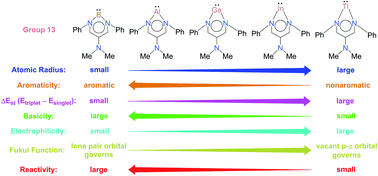
Three fundamental concepts (aromaticity/basicity/electrophilicity), being heavily used in modern chemistry, have been applied in this work to study the chemical reactivity of six-membered-ring group 13 N-heterocyclic carbenes (G13-6-Rea; G13 = group 13 elements) using density functional theory (BP86-D3(BJ)/def2-TZVP). G13-6-Rea is isolobal to benzene. Two model reactions have been used in the present study: the insertion reaction of G13-6-Rea with methane and the [1 + 2] cycloaddition reaction of G13-6-Rea with ethene. Our theoretical analysis reveals that the chemical reactivity of B-6-Rea, Al-6-Rea, and Ga-6-Rea is governed by their HOMO (the sp2-σ lone pair orbital on the G13 element), and thus they can be considered nucleophiles. Conversely, the chemical behavior of In-6-Rea and Tl-6-Rea is determined by their LUMO (the vacant p-π orbital on the G13 element), and thus they can be considered electrophiles. On the basis of the VBSCD (valence bond state correlation diagram) model and ASM (activation strain model), this theoretical evidence demonstrates that the origin of activation barriers for the above model reactions is due to the atomic radius of the pivotal group 13 element in the six-membered-ring of G13-6-Rea. Accordingly, our theoretical conclusions suggest that the lower the atomic number and the smaller the atomic radius of the G13 atom, the higher the aromaticity of the six-membered-ring of G13-6-Rea and the smaller the singlet–triplet energy splitting ΔEst of this N-heterocyclic carbene analogue, which will facilitate its chemical reactions. The theoretical findings originating from this study allow many predictions in experiments to be made.
 Please wait while we load your content...
Please wait while we load your content...

