
An efficient approach to study membrane nano-inclusions: from the complex biological world to a simple representation
 *a
*a
Abstract
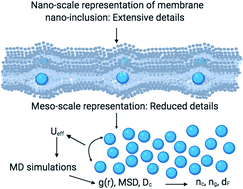
Membrane nano-inclusions (NIs) are of great interest in biophysics, materials science, nanotechnology, and medicine. We hypothesized that the NIs within a biological membrane bilayer interact via a simple and efficient interaction potential, inspired by previous experimental and theoretical work. This interaction implicitly treats the membrane lipids but takes into account its effect on the NIs micro-arrangement. Thus, the study of the NIs is simplified to a two-dimensional colloidal system with implicit solvent. We calculated the structural properties from Molecular Dynamics simulations (MD), and we developed a Scaling Theory to discuss their behavior. We determined the thermal properties through potential energy per NI and pressure, and we discussed their variation as a function of the NIs number density. We performed a detailed study of the NIs dynamics using two approaches, MD simulations, and Dynamics Theory. We identified two characteristic values of number density, namely a critical number density nc = 3.67 × 10−3 Å−2 corresponded to the apparition of chain-like structures along with the liquid dispersed structure and the gelation number density ng = 8.40 × 10−3 Å−2 corresponded to the jamming state. We showed that the aggregation structure of NIs is of fractal dimension dF < 2. Also, we identified three diffusion regimes of membrane NIs, namely, normal for n < nc, subdiffusive for nc ≤ n < ng, and blocked for n ≥ ng. Thus, this paper proposes a simple and effective approach for studying the physical properties of membrane NIs. In particular, our results identify scaling exponents related to the microstructure and dynamics of membrane NIs.
 Please wait while we load your content...
Please wait while we load your content...

