
Accurate location of hydrogen atoms in hydrogen bonds of tizoxanide from the combination of experimental and theoretical models†

Abstract
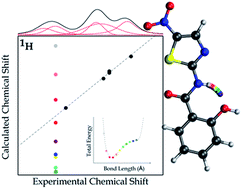
To obtain detailed information about the position of hydrogen atoms in hydrogen bonds, HBs, of crystalline organic molecular compounds is not an easy task. In this work we propose a combination of ssNMR experimental data with theoretical procedures to get such information. Furthermore, the combination of experimental and theoretical models provides us with well-defined grounds to analyse the strength of π-stacking interactions between layers of hydrogen bonded molecules. Two different theoretical models were considered, both approaches being quite different. The first one is a solid-state model, so that the periodicity of a crystalline system underlies calculations of the electronic energy, the electronic density and NMR parameters. The other one is a molecular model in which molecules are taken as isolated monomers, dimers and tetramers. These two models were applied to the tizoxanide, TIZ, molecular crystal though it can widely be applied to any other molecular crystal. By the application of the quantum molecular model it was possible to learn about the way the intermolecular HBs affect the position of hydrogen atoms that belong to HBs in TIZ. This molecule has two intermolecular HBs that stabilize the structure of a basic dimer, but it also has an intramolecular HB in each monomer whose position should be optimized together with the other ones. We found that by doing this it is possible to obtain reliable results of calculations of NMR spectroscopic parameters. Working with the solid-state model we found that any local variation of the TIZ crystalline structure is correlated with the variation of the values of the NMR parameters of each nucleus. The excellent agreement between experimental and calculated chemical shifts leads to the conclusion that the N10–H10 bond distance should be (1.00 ± 0.02) Å.
- This article is part of the themed collection: ENQA - 20th Brazilian Meeting on Analytical Chemistry
 Please wait while we load your content...
Please wait while we load your content...

