
High-efficient ab initio Bayesian active learning method and applications in prediction of two-dimensional functional materials†

Abstract
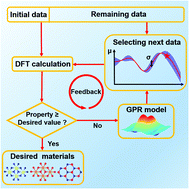
Beyond the conventional trial-and-error method, machine learning offers a great opportunity to accelerate the discovery of functional materials, but still often suffers from difficulties such as limited materials data and the unbalanced distribution of target properties. Here, we propose the ab initio Bayesian active learning method that combines active learning and high-throughput ab initio calculations to accelerate the prediction of desired functional materials with ultrahigh efficiency and accuracy. We apply it as an instance to a large family (3119) of two-dimensional hexagonal binary compounds with unbalanced materials properties, and accurately screen out the materials with maximal electric polarization and proper photovoltaic band gaps, respectively, whereas the computational costs are significantly reduced by only calculating a few tenths of the possible candidates in comparison with a random search. This approach shows the enormous advantages for the cases with unbalanced distribution of target properties. It can be readily applied to seek a broad range of advanced materials.
- This article is part of the themed collection: 2021 Nanoscale HOT Article Collection
 Please wait while we load your content...
Please wait while we load your content...

