
Flexoelectricity in atomic monolayers from first principles†

Abstract
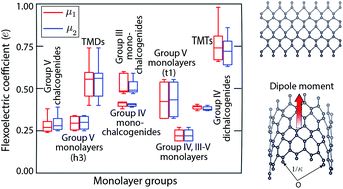
We study the flexoelectric effect in fifty-four select atomic monolayers using ab initio Density Functional Theory (DFT). Specifically, considering representative materials from each of the Group III monochalcogenides, transition metal dichalcogenides (TMDs), Groups IV, III–V, and V monolayers, Group IV dichalcogenides, Group IV monochalcogenides, transition metal trichalcogenides (TMTs), and Group V chalcogenides, we perform symmetry-adapted DFT simulations to calculate transversal flexoelectric coefficients along the principal directions at practically relevant bending curvatures. We find that the materials demonstrate linear behavior and have similar coefficients along both principal directions, with values for TMTs being up to a factor of five larger than those of graphene. In addition, we find electronic origins for the flexoelectric effect, which increases with monolayer thickness, elastic modulus along the bending direction, and sum of polarizability of constituent atoms.
- This article is part of the themed collection: Editor’s Choice: Computational studies of nanomaterials for energy, catalysis and electronics
 Please wait while we load your content...
Please wait while we load your content...

