
The size-dependent influence of palladium doping on the structures of cationic gold clusters†
 *a
Laia
Delgado-Callico,
b
Gao-Lei
Hou,
‡a
Francesca
Baletto,
b
Joost M.
Bakker
c
and
Ewald
Janssens
a
*a
Laia
Delgado-Callico,
b
Gao-Lei
Hou,
‡a
Francesca
Baletto,
b
Joost M.
Bakker
c
and
Ewald
Janssens
a
Abstract
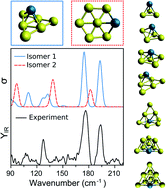
The physicochemical properties of small metal clusters strongly depend on their precise geometry. Determining such geometries, however, is challenging, particularly for clusters formed by multiple elements. In this work, we combine infrared multiple photon dissociation spectroscopy and density functional theory calculations to investigate the lowest-energy structures of Pd doped gold clusters, PdAun−1+ (n ≤ 10). The high-quality experimental spectra allow for an unambiguous determination of the structures adopted by the clusters. Our results show that the Pd–Au interaction is so large that the structures of PdAun−1+ and Aun+ are very different. Pd doping induces a 2D to 3D transition at much smaller cluster sizes than for pure Aun+ clusters. PdAun−1+ clusters are three-dimensional from n = 4, whereas for Aun+ this transition only takes place at n = 7. Despite the strong Au–Pd interaction, the Aun−1+ cluster geometries remain recognizable in PdAun−1+ up to n = 7. This is particularly clear for PdAu6+. In PdAu8+ and PdAu9+, Pd triggers major rearrangements of the Au clusters, which adopt pyramidal shapes. For PdAu4+ we find a geometry that was not considered in previous studies, and the geometry found for PdAu8+ does not correspond to the lowest-energy structure predicted by DFT, suggesting kinetic trapping during formation. This work demonstrates that even with the continuous improvement of computational methods, unambiguous assignment of cluster geometries still requires a synergistic approach, combining experiment and computational modelling.
 Please wait while we load your content...
Please wait while we load your content...

