
First principles study of the stability of MXenes under an electron beam†

Abstract
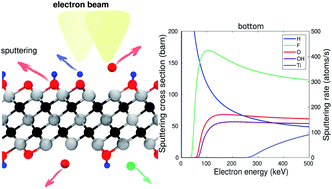
Interactions between two-dimensional MXene sheets and electron beams of a (scanning) transmission electron microscope are studied by first-principles calculations. We simulated the knock-on sputtering threshold for Ti3C2 MXene sheets via ab initio molecular dynamics simulations and for five other MXenes (Ti2C, Ti2N, Nb2C, Mo2TiC2, and Ti3CN) approximately from defect formation energies. We evaluated the sputtering cross section and sputtering rates and based on those evaluated the surface composition. We find that at the exit surface and for “low” TEM energies H and F sputter at equal rates, but at “high” TEM energies the F is sputtered most strongly. In the entry surface, H sputtering dominates. The results were found to be largely similar for all studied MXenes, and although the sputtering thresholds varied between the different metal atoms the thresholds were always too high to lead to significant sputtering of the metal atoms. We simulated electron microscope images at the successive stages of sputtering and found that while it is likely difficult to identify surface groups based on the spot intensities, the local contraction of the lattice around O groups should be observable. We also studied MXenes encapsulated with graphene and found them to provide efficient protection from knock-on damage for all surface group atoms except H.
 Please wait while we load your content...
Please wait while we load your content...

