
Density functional theory study of thiophene desulfurization and conversion of desulfurization products on the Ni(111) surface and Ni55 cluster: implication for the mechanism of reactive adsorption desulfurization over Ni/ZnO catalysts†

Abstract
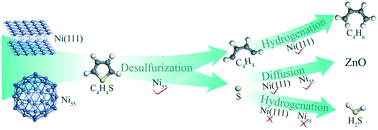
Ni/ZnO catalysts have been well recognized by industry and academia for exhibiting excellent desulfurization activities. However, the intrinsic reaction mechanism on the Ni active center is still obscure. Herein, we performed periodic density functional theory (DFT) calculations to study thiophene desulfurization and conversion of desulfurization products on the Ni(111) surface and Ni55 cluster, and clarify the size effect of the Ni substrate and the essential role of hydrogen. The thiophene molecule binds more strongly to Ni55 than Ni(111), and proceeds easily along the direct desulfurization pathway without prior hydrogenation on both Ni(111) and Ni55. Ni55 exhibits higher desulfurization activity while Ni(111) performs better in converting the remaining C4H4 species to butadiene and the deposited S atom to H2S. In contrast to the classic S transfer mechanism via H2S, we found that direct S diffusion occurs easily on the Ni substrate and has priority over its further hydrogenation to H2S, indicating that the S diffusion mechanism could play an important role in transferring surface S from Ni to ZnO. The results also show that the C–S bond rupture of thiophene and subsequent S removal from Ni tend to proceed without the assistance of hydrogen, and hydrogen mainly takes part in the hydrogenation of C4H4 species. The present work clearly demonstrates that the rate-determining step for thiophene desulfurization and subsequent alkene formation is the C4H4 hydrogenation rather than the cleavage of C–S bonds, in accordance with the experimental results.
 Please wait while we load your content...
Please wait while we load your content...

