
Site-selective modification strategies in antibody–drug conjugates
 †ab
Jonathan D.
Bargh,
†a
Friederike M.
Dannheim,
†a
Hikaru
Seki,
†a
Andrew J.
Counsell,
†a
Xiaoxu
Ou,
†a
Elaine
Fowler,
†a
Nicola
Ashman,
†a
Yuri
Takada,
†a
Albert
Isidro-Llobet,
c
Jeremy S.
Parker,
d
Jason S.
Carroll
b
and
David R.
Spring
*a
†ab
Jonathan D.
Bargh,
†a
Friederike M.
Dannheim,
†a
Hikaru
Seki,
†a
Andrew J.
Counsell,
†a
Xiaoxu
Ou,
†a
Elaine
Fowler,
†a
Nicola
Ashman,
†a
Yuri
Takada,
†a
Albert
Isidro-Llobet,
c
Jeremy S.
Parker,
d
Jason S.
Carroll
b
and
David R.
Spring
*a
Abstract
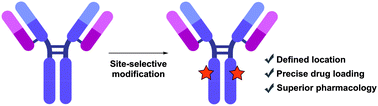
Antibody–drug conjugates (ADCs) harness the highly specific targeting capabilities of an antibody to deliver a cytotoxic payload to specific cell types. They have garnered widespread interest in drug discovery, particularly in oncology, as discrimination between healthy and malignant tissues or cells can be achieved. Nine ADCs have received approval from the US Food and Drug Administration and more than 80 others are currently undergoing clinical investigations for a range of solid tumours and haematological malignancies. Extensive research over the past decade has highlighted the critical nature of the linkage strategy adopted to attach the payload to the antibody. Whilst early generation ADCs were primarily synthesised as heterogeneous mixtures, these were found to have sub-optimal pharmacokinetics, stability, tolerability and/or efficacy. Efforts have now shifted towards generating homogeneous constructs with precise drug loading and predetermined, controlled sites of attachment. Homogeneous ADCs have repeatedly demonstrated superior overall pharmacological profiles compared to their heterogeneous counterparts. A wide range of methods have been developed in the pursuit of homogeneity, comprising chemical or enzymatic methods or a combination thereof to afford precise modification of specific amino acid or sugar residues. In this review, we discuss advances in chemical and enzymatic methods for site-specific antibody modification that result in the generation of homogeneous ADCs.
 Please wait while we load your content...
Please wait while we load your content...

