
A decomposition mechanism for Mn2(DSBDC) metal–organic frameworks in the presence of water molecules†

Abstract
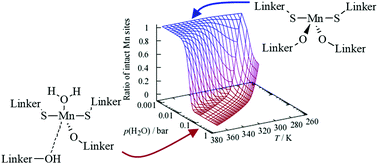
In this work, we investigate the effects of water on the structural stability of Mn2(DSBDC) metal–organic framework (MOF) using DFT-based calculations. It has been found that the adsorption of multiple water molecules forming a hydrogen bond network around the Mn centers plays an important role in the decomposition process. Different effects contribute to the destabilization of the MOF: water molecules that directly coordinate to the open sites displayed by a part of the Mn centers can induce a significant shift in the charge distribution as indicated by the analysis of charge density differences and the Bader charges. This adsorption process leads to a slight elongation of the metal-linker bonds. The direct interaction with the Mn center is the most stable adsorption mode for water in Mn2(DSBDC). Once these adsorption sites at the Mn centers are fully occupied, additional water molecules start to bind via hydrogen bonds to the already present water molecules or, more importantly, to the linker molecules. This, in return, leads to a significant weakening of the Mn-linker bonds, thus allowing water insertion into the Mn-linker bonds with a barrier of only 0.16 eV, which is believed to initiate the decomposition of the Mn2(DSBDC) framework. Based on a kinetic Monte Carlo model, it can be shown that high temperatures can prevent the adsorption of water molecules around the Mn sites and thus slow down the MOF decomposition.
 Please wait while we load your content...
Please wait while we load your content...

