
Molecular excited state calculations with adaptive wavefunctions on a quantum eigensolver emulation: reducing circuit depth and separating spin states†

Abstract
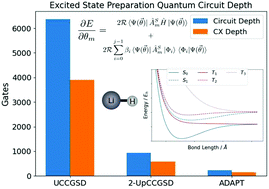
Ab initio electronic excited state calculations are necessary for the quantitative study of photochemical reactions, but their accurate computation on classical computers is plagued by prohibitive resource scaling. The Variational Quantum Deflation (VQD) is an extension of the quantum-classical Variational Quantum Eigensolver (VQE) algorithm for calculating electronic excited state energies, and has the potential to address some of these scaling challenges using quantum computers. However, quantum computers available in the near term can only support a limited number of quantum circuit operations, so reducing the quantum computational cost in VQD methods is critical to their realisation. In this work, we investigate the use of adaptive quantum circuit growth (ADAPT-VQE) in excited state VQD calculations, a strategy that has been successful previously in reducing the resources required for ground state energy VQE calculations. We also invoke spin restrictions to separate the recovery of eigenstates with different spin symmetry to reduce the number of calculations and accumulation of errors in computing excited states. We created a quantum eigensolver emulation package - Quantum Eigensolver Building on Achievements of Both quantum computing and quantum chemistry (QEBAB) – for testing the proposed adaptive procedure against two existing VQD methods that use fixed-length quantum circuits: UCCGSD-VQD and k-UpCCGSD-VQD. For a lithium hydride test case we found that the spin-restricted adaptive growth variant of VQD uses the most compact circuits out of the tested methods by far, consistently recovers adequate electron correlation energy for different nuclear geometries and eigenstates while isolating the singlet and triplet manifold. This work is a further step towards developing techniques which improve the efficiency of hybrid quantum algorithms for excited state quantum chemistry, opening up the possibility of exploiting real quantum computers for electronic excited state calculations sooner than previously anticipated.
- This article is part of the themed collection: Quantum Computing and Quantum Information Storage
 Please wait while we load your content...
Please wait while we load your content...

