
A combined spectroscopic and computational investigation on dispersion-controlled docking of Ar atoms on 2-(2′-pyridyl)benzimidazole†
 a
and
Surajit
Maity
*a
a
and
Surajit
Maity
*a
Abstract
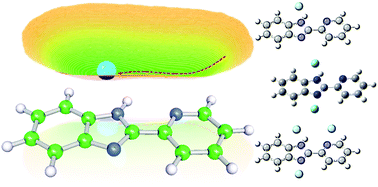
The dispersion-controlled docking of inert Ar atoms on the face of polycyclic 2-(2′-pyridyl)-benzimidazole (PBI) was studied experimentally aided by computational findings. The PBI–Arn (n = 1–3) complexes were produced in a supersonically jet-cooled molecular beam and probed using resonant two-photon ionization coupled with a time-of-flight mass spectrometric detection scheme and laser-induced fluorescence spectroscopy. The ground state vibrational frequencies were obtained from single vibronic level fluorescence spectroscopy. The formation of multiple isomers was verified using UV-UV hole-burning spectroscopy. The geometries of PBI–Arn (n = 1–3) complexes were derived by mutual agreement between experimental findings and computational results such as vibrational frequencies in the ground and excited electronic states, Franck–Condon factors and spectral shift of the S1 ← S0 transitions. All the above analyses provided good agreement between the experimental and simulated spectrum with the most stable PBI–Arn (n = 1–3) clusters. The highest intermolecular interaction between PBI and Ar was obtained with the Ar atom positioned above the imidazolyl ring. A second Ar atom was preferably docking on the other side of the imidazolyl ring than the pyridyl ring. The subsequent addition of the third Ar atom preferred the position above the pyridyl ring. The current investigation can be useful to investigate the preferential docking of dispersion-controlled interacting partners in multifunctional aromatic side chains present in biological systems.
 Please wait while we load your content...
Please wait while we load your content...

