
cis → trans photoisomerisation of azobenzene: a fresh theoretical look†

Abstract
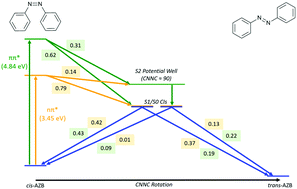
The cis → trans photo-isomerisation mechanism of azobenzene, after excitation to the nπ* and ππ* states, is revisited using high-level ab initio surface hopping mixed quantum-classical dynamics in combination with multi-reference CASSCF electronic structure calculations. A reduction of photoisomerisation quantum yield of 0.10 on exciting to the higher energy ππ* state compared to the lower energy nπ* state is obtained, in close agreement with the most recent experimental values [Ladányi et al., Photochem. Photobiol. Sci., 2017, 16, 1757–1761] which re-examined previous literature values which showed larger changes in quantum yield. By direct comparison of both excitations, we have found that the explanation for the decrease in quantum yield is not the same as for the reduction observed in the trans → cis photoisomerisation. In contrast to the trans → cis scenario, S1 → S0 decay does not occur at ‘earlier’ C–N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–C angles along the central torsional coordinate after ππ* excitation, as in the cis → trans case the rotation about this coordinate occurs too rapidly. The wavelength dependency of the quantum yield is instead found to be due to a potential well on the S2 surface, from which either cis or trans-azobenzene can be formed. While this well is accessible after both excitations, it is more easily accessed after ππ* excitation – an additional 15–17% of photochromes, which under nπ* excitation would have exclusively formed the trans isomer, are trapped in this well after ππ* excitation. The probability of forming the cis isomer when leaving this well is also higher after ππ* excitation, increasing from 9% to 35%. The combination of these two factors results in the reduction of 0.10 of the quantum yield of photoisomerisation on ππ* excitation of cis-azobenzene, compared to nπ* excitation.
N–C angles along the central torsional coordinate after ππ* excitation, as in the cis → trans case the rotation about this coordinate occurs too rapidly. The wavelength dependency of the quantum yield is instead found to be due to a potential well on the S2 surface, from which either cis or trans-azobenzene can be formed. While this well is accessible after both excitations, it is more easily accessed after ππ* excitation – an additional 15–17% of photochromes, which under nπ* excitation would have exclusively formed the trans isomer, are trapped in this well after ππ* excitation. The probability of forming the cis isomer when leaving this well is also higher after ππ* excitation, increasing from 9% to 35%. The combination of these two factors results in the reduction of 0.10 of the quantum yield of photoisomerisation on ππ* excitation of cis-azobenzene, compared to nπ* excitation.
- This article is part of the themed collections: PCCP 2021 Emerging Investigators and 2021 PCCP HOT Articles
 Please wait while we load your content...
Please wait while we load your content...

