
Gaseous cyclodextrin-closo-dodecaborate complexes χCD·B12X122− (χ = α, β, and γ; X = F, Cl, Br, and I): electronic structures and intramolecular interactions†
 b
Wenjin
Cao,
b
Markus
Rohdenburg,
cd
Marc C.
Nierstenhöfer,
e
Zhipeng
Li,
a
Yan
Yang,
a
Carsten
Jenne,
e
Jonas
Warneke,
cg
Haitao
Sun,
*ah
Xue-Bin
Wang
*b
b
Wenjin
Cao,
b
Markus
Rohdenburg,
cd
Marc C.
Nierstenhöfer,
e
Zhipeng
Li,
a
Yan
Yang,
a
Carsten
Jenne,
e
Jonas
Warneke,
cg
Haitao
Sun,
*ah
Xue-Bin
Wang
*b
Abstract
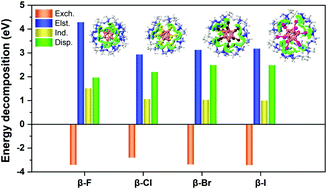
A fundamental understanding of cyclodextrin-closo-dodecaborate inclusion complexes is of great interest in supramolecular chemistry. Herein, we report a systematic investigation on the electronic structures and intramolecular interactions of perhalogenated closo-dodecaborate dianions B12X122− (X = F, Cl, Br and I) binding to α-, β-, and γ-cyclodextrins (CDs) in the gas phase using combined negative ion photoelectron spectroscopy (NIPES) and density functional theory (DFT) calculations. The vertical detachment energy (VDE) of each complex and electronic stabilization of each dianion due to the CD binding (ΔVDE, relative to the corresponding isolated B12X122−) are determined from the experiments along α-, β- and γ-CD in the form of VDE (ΔVDE): 4.00 (2.10), 4.33 (2.43), and 4.30 (2.40) eV in X = F; 4.09 (1.14), 4.64 (1.69), and 4.69 (1.74) eV in X = Cl; 4.11 (0.91), 4.58 (1.38), and 4.70 (1.50) eV in X = Br; and 3.54 (0.74), 3.88 (1.08), and 4.05 (1.25) eV in X = I, respectively. All complexes have significantly higher VDEs than the corresponding isolated dodecaborate dianions with ΔVDE spanning from 0.74 eV at (α, I) to 2.43 eV at (β, F), sensitive to both host CD size and guest substituent X. DFT-optimized complex structures indicate that all B12X122− prefer binding to the wide openings of CDs with the insertion depth and binding motif strongly dependent on the CD size and halogen X. Dodecaborate anions with heavy halogens, i.e., X = Cl, Br, and I, are found outside of α-CD, while B12F122− is completely wrapped by γ-CD. Partial embedment of B12X122− into CDs is observed for the other complexes via multipronged B–X⋯H–O/C interlocking patterns. The simulated spectra based on the density of states agree well with those of the experiments and the calculated VDEs well reproduce the experimental trends. Molecular orbital analyses suggest that the spectral features at low binding energies originated from electrons detached from the dodecaborate dianion, while those at higher binding energies are derived from electron detachment from CDs. Energy decomposition analyses reveal that the electrostatic interaction plays a dominating role in contributing to the host–guest interactions for the X = F series partially due to the formation of a O/C–H⋯X–B hydrogen bonding network, and the dispersion forces gradually become important with the increase of halogen size.
 Please wait while we load your content...
Please wait while we load your content...

