
Oxidative etching of S-vacancy defective MoS2 monolayer upon reaction with O2†

Abstract
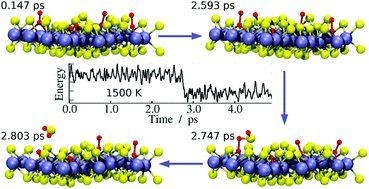
The reactions of O2 with S vacancy sites within a MoS2 monolayer were investigated using density functional theory calculations. We considered the following defects: single S vacancy, double S vacancy, two adjacent S vacancies and two S vacancies separated by a sulphur atom. We found that the surface distribution of S vacancy sites plays a key role in determining the surface reactivity towards O2. We observed the desorption of SO2 only for the last vacancy distribution. For the other cases, the surface becomes passivated with very stable structures having O atoms on the original vacancy sites and in some cases an SO group in an adjacent position. The ab initio molecular dynamics simulations showed that the impingement of the O2 molecule on an S vacancy site produces a stable chemisorbed O2 molecule with an upright configuration. The surface reactions initiate after the O2 molecule switches to the lying-down configuration which favours the breakage of the O–O bond and the concurrent formation of S–O bonds. In the most reactive vacancy site configuration, the dissociation of the first O2 molecule produces an SO intermediate which finally leads to desorption of SO2 after oxygen abstraction from the other adjacent O2 molecule. The formation of a MoO3 moiety within the monolayer was also observed in the molecular dynamic simulations at higher oxidation levels.
- This article is part of the themed collection: 2021 PCCP HOT Articles
 Please wait while we load your content...
Please wait while we load your content...

