
Accurate and rapid prediction of pKa of transition metal complexes: semiempirical quantum chemistry with a data-augmented approach†
 *a
Evgeny A.
Pidko
*a
*a
Evgeny A.
Pidko
*a
Abstract
Rapid and accurate prediction of reactivity descriptors of transition metal (TM) complexes is a major challenge for contemporary quantum chemistry. The recently-developed GFN2-xTB method based on the density functional tight-binding theory (DFT-B) is suitable for high-throughput calculation of geometries and thermochemistry for TM complexes albeit with moderate accuracy. Herein we present a data-augmented approach to improve substantially the accuracy of the GFN2-xTB method for the prediction of thermochemical properties using pKa values of TM hydrides as a representative model example. We constructed a comprehensive database for ca. 200 TM hydride complexes featuring the experimentally measured pKa values as well as the GFN2-xTB-optimized geometries and various computed electronic and energetic descriptors. The GFN2-xTB results were further refined and validated by DFT calculations with the hybrid PBE0 functional. Our results show that although the GFN2-xTB performs well in most cases, it fails to adequately describe TM complexes featuring multicarbonyl and multihydride ligand environments. The dataset was analyzed with the ordinary least squares (OLS) fitting and was used to construct an automated machine learning (AutoML) approach for the rapid estimation of pKa of TM hydride complexes. The results obtained show a high predictive power of the very fast AutoML model (RMSE ∼ 2.7) comparable to that of the much slower DFT calculations (RMSE ∼ 3). The presented data-augmented quantum chemistry-based approach is promising for high-throughput computational screening workflows of homogeneous TM-based catalysts.
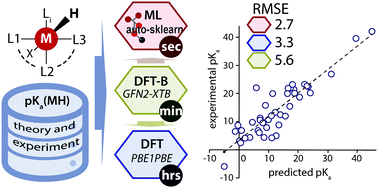
- This article is part of the themed collections: Emerging AI Approaches in Physical Chemistry and Quantum Theory: The Challenge of Transition Metal Complexes
 Please wait while we load your content...
Please wait while we load your content...

