
Accelerating atomistic simulations with piecewise machine-learned ab Initio potentials at a classical force field-like cost†
 *a
*a
Abstract
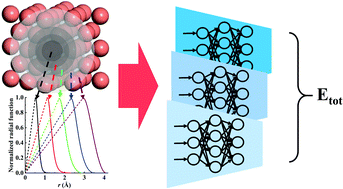
Recently, machine learning methods have become easy-to-use tools for constructing high-dimensional interatomic potentials with ab initio accuracy. Although machine-learned interatomic potentials are generally orders of magnitude faster than first-principles calculations, they remain much slower than classical force fields, at the price of using more complex structural descriptors. To bridge this efficiency gap, we propose an embedded atom neural network approach with simple piecewise switching function-based descriptors, resulting in a favorable linear scaling with the number of neighbor atoms. Numerical examples validate that this piecewise machine-learning model can be over an order of magnitude faster than various popular machine-learned potentials with comparable accuracy for both metallic and covalent materials, approaching the speed of the fastest embedded atom method (i.e. several μs per atom per CPU core). The extreme efficiency of this approach promises its potential in first-principles atomistic simulations of very large systems and/or in a long timescale.
- This article is part of the themed collection: Emerging AI Approaches in Physical Chemistry
 Please wait while we load your content...
Please wait while we load your content...

