
Ab initio Gibbs ensemble Monte Carlo simulations of the liquid–vapor equilibrium and the critical point of sodium
 *a
François
Soubiran
ac
and
*a
François
Soubiran
ac
and
Abstract
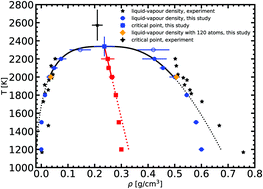
The ab initio (ai) Gibbs ensemble (GE) Monte Carlo (MC) method coupled with Kohn–Sham density functional theory is successful in predicting the liquid–vapour equilibrium of insulating systems. Here we show that the aiGEMC method can be used to study also metallic systems, where the excited electronic states play an important role and cannot be neglected. For this we include the electronic free energy in the formulation of the effective energy of the system to be used in the acceptance criteria for the MC moves. The application of this aiGEMC method to sodium yields a good agreement with available experimental data on the liquid–vapour equilibrium densities. We predict a critical point for sodium at 2338 ± 108 K and 0.24 ± 0.03 g cm−3. The liquid structure stemming from aiGEMC simulations is very similar to the one from ab initio molecular dynamics. Since this method can determine phase transition without computing the Gibbs free energy, it may offer a new possibility to study other materials with a reasonable computational cost.
 Please wait while we load your content...
Please wait while we load your content...

