
Geometrical and energetic characteristics of Se⋯Se interactions in crystal structures of organoselenium molecules†

Abstract
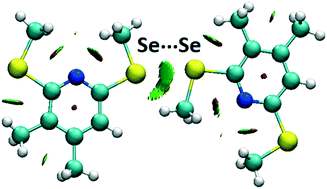
Non-covalent selenium–selenium interactions between selenium-containing organic molecules were studied in crystal structures from the Cambridge Structural Database and by high-level quantum chemical calculations. Se⋯Se contacts in the crystal structures were analyzed, and the most frequent patterns were identified and used to design the model systems for quantum chemical calculations. The strongest calculated Se⋯Se interaction (ΔECCSD(T)/CBS = −2.31 kcal mol−1) was identified in the model system with a mutual parallel orientation of interacting molecules. In the crystal structures, this orientation of molecules is predominant. In the geometry with the σ-hole bonding, the interaction is somewhat weaker (ΔECCSD(T)/CBS = −2.13 kcal mol−1). NCI analysis showed that Se⋯Se interaction in the most stable geometries is further enhanced by hydrogen bonding of Se–H⋯Se or C–H⋯Se type. The results of energy decomposition analysis (SAPT) calculations revealed that the nature of the Se⋯Se interaction is predominantly dispersive with a strong electrostatic contribution. The results of the energy decomposition analysis also suggest that the electrostatic component has a crucial role in defining the geometry of selenium–selenium interactions due to their directional nature.
 Please wait while we load your content...
Please wait while we load your content...

