
Computer modeling of polymer stars in variable solvent conditions: a comparison of MD simulations, self-consistent field (SCF) modeling and novel hybrid Monte Carlo SCF approach

Abstract
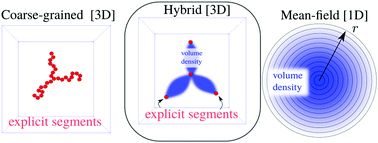
Computer-aided modeling is a systematic approach to grasp the physics of macromolecules, but it remains essential to know when to trust the results and when not. For a polymer star, we consider three approaches: (i) Molecular Dynamics (MD) simulations and implementing a coarse-grained model, (ii) the self-consistent field approach based on a mean-field approximation and implementing the lattice model due to Scheutjens and Fleer (SF-SCF) and (iii) novel hybrid Monte Carlo self-consistent field (MC-SCF) method, which combines a coarse-grained model driven by a Monte Carlo method and a mean-field representation driven by SF-SCF. We compare the performance of these approaches under a wide range of solvent qualities. The MD approach is formally the most exact but suffers from reasonable convergence. The mean-field approach works similarly in all solvent qualities but is quantitatively least accurate. The MC-SCF hybrid allows us to combine the benefits of the simulation route and the effective performance of SCF. We consider the center-to-end distance Rce, the radius of gyration Rg2 of the star and the polymer density profiles φ(r) of polymer-segments in it. All three methods show a good qualitative agreement one to another. The MC-SCF method is in good agreement with the scaling predictions in the whole range of solvent quality values showing that it grasps the essential physics while remaining computationally in bounds.
 Please wait while we load your content...
Please wait while we load your content...

