
Alchemical absolute protein–ligand binding free energies for drug design†

Abstract
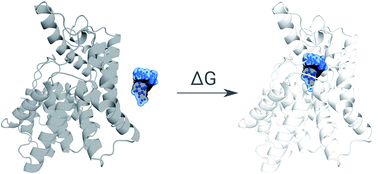
The recent advances in relative protein–ligand binding free energy calculations have shown the value of alchemical methods in drug discovery. Accurately assessing absolute binding free energies, although highly desired, remains a challenging endeavour, mostly limited to small model cases. Here, we demonstrate accurate first principles based absolute binding free energy estimates for 128 pharmaceutically relevant targets. We use a novel rigorous method to generate protein–ligand ensembles for the ligand in its decoupled state. Not only do the calculations deliver accurate protein–ligand binding affinity estimates, but they also provide detailed physical insight into the structural determinants of binding. We identify subtle rotamer rearrangements between apo and holo states of a protein that are crucial for binding. When compared to relative binding free energy calculations, obtaining absolute binding free energies is considerably more challenging in large part due to the need to explicitly account for the protein in its apo state. In this work we present several approaches to obtain apo state ensembles for accurate absolute ΔG calculations, thus outlining protocols for prospective application of the methods for drug discovery.
- This article is part of the themed collection: Computational protein design and structure prediction: Celebrating the 2024 Nobel Prize in Chemistry
 Please wait while we load your content...
Please wait while we load your content...

