
Machine learning dielectric screening for the simulation of excited state properties of molecules and materials†

Abstract
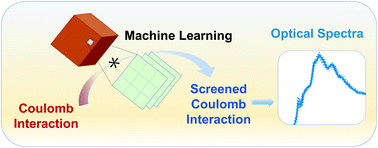
Accurate and efficient calculations of absorption spectra of molecules and materials are essential for the understanding and rational design of broad classes of systems. Solving the Bethe–Salpeter equation (BSE) for electron–hole pairs usually yields accurate predictions of absorption spectra, but it is computationally expensive, especially if thermal averages of spectra computed for multiple configurations are required. We present a method based on machine learning to evaluate a key quantity entering the definition of absorption spectra: the dielectric screening. We show that our approach yields a model for the screening that is transferable between multiple configurations sampled during first principles molecular dynamics simulations; hence it leads to a substantial improvement in the efficiency of calculations of finite temperature spectra. We obtained computational gains of one to two orders of magnitude for systems with 50 to 500 atoms, including liquids, solids, nanostructures, and solid/liquid interfaces. Importantly, the models of dielectric screening derived here may be used not only in the solution of the BSE but also in developing functionals for time-dependent density functional theory (TDDFT) calculations of homogeneous and heterogeneous systems. Overall, our work provides a strategy to combine machine learning with electronic structure calculations to accelerate first principles simulations of excited-state properties.
- This article is part of the themed collections: Materials & Energy in Chemical Science - most popular articles 2021 and Editor’s Choice – Graeme Day
 Please wait while we load your content...
Please wait while we load your content...

